ABSTRACT
CUBAP: An Interactive Web Portal for Analyzing Codon Usage Bias Across Populations
Although synonymous codon usage does not change the protein primary structure, codon choice significantly impacts translational
and transcriptional efficiency, gene expression, the secondary structure of both mRNA and proteins, and has been implicated in
various diseases. While codon usage has been widely studied, population-specific differences in codon usage biases remain largely
unexplored. Here, we present a web server, https://cubap.byu.edu, to facilitate analyses of codon usage biases across populations
(CUBAP). Using data from the 1000 Genomes Project, we calculated codon frequencies, codon aversion, identical codon pairing,
co-tRNA codon pairing, ramp sequences, and nucleotide composition including GC content in 17,638 genes. We present comprehensive
comparisons of these codon usage biases across 2,504 individuals spanning 26 subpopulations in five superpopulations and
visually depict these differences online using Microsoft Power BI. We found that codon pairing significantly differs between
populations in 46% of genes, allowing us to successfully predict the place of origin for African and East Asian individuals
using only codon pairing biases with 98.8% and 100% accuracy, respectively. We also performed a case study using CUBAP to
analyze population-specific effects of synonymous variant rs10065172, which is associated with Crohn's disease in European
populations but not Japanese or African populations. We propose that a significant bias toward decreased CTG pairing in the
immunity related GTPase M (IRGM) gene in East Asian and African populations may contribute to the decreased association
of rs10065172 with Crohn's disease within these populations. CUBAP displays the most comprehensive analysis of codon usage
biases across human populations to date. It facilitates in-depth gene-specific and codon-specific visualization that will
aid in analyzing candidate genes identified in genome-wide association studies, identifying functional implications of
synonymous variants, predicting population-specific impacts of synonymous variants, and categorizing genetic biases unique to certain populations.
Populations Included from the 1000 Genomes Project
| Superpopulation |
Subpopulation |
Description |
| Africa |
ASW |
African Ancestry in Southwest US |
| ACB |
African Caribbean in Barbados |
| ESN |
Esan in Nigeria |
| GWD |
Gambian in Western Division, The Gambia |
| LWK |
Luhya in Webuye, Kenya |
| MSL |
Mende in Sierra Leone |
| YRI |
Yoruba in Ibadan, Nigeria |
| America |
CLM |
Colombian in Medellin, Colombia |
| MXL |
Mexican Ancestry in Los Angeles, California |
| PEL |
Peruvian in Lima, Peru |
| PUR |
Puerto Rican in Puerto Rico |
| East Asia |
CDX |
Chinese Dai in Xishuangbanna, China |
| CHB |
Han Chinese in Bejing, China |
| CHS |
Southern Han Chinese, China |
| JPT |
Japanese in Tokyo, Japan |
| KHV |
Kinh in Ho Chi Minh City, Vietnam |
| Europe |
CEU |
Utah residents with Northern and Western European ancestry |
| GBR |
British in England and Scotland |
| FIN |
Finnish in Finland |
| IBS |
Iberian populations in Spain |
| TSI |
Toscani in Italy |
| South Asia |
BEB |
Bengali in Bangladesh |
| GIH |
Gujarati Indian in Houston,TX |
| ITU |
Indian Telugu in the UK |
| PJL |
Punjabi in Lahore,Pakistan |
| STU |
Sri Lankan Tamil in the UK |
RESULTS
What did we find?
A more detailed look at the differences between these specific genes can expand our knowledge of the extent of their fitness and differences. Data such as codon usage bias and codon pairing, both key aspects of protein production, will be extremely useful, especially when studying genes that produce important proteins such as insulin. It has been observed that an organism’s codon bias, codon pairing, and codon aversion are all related to its location in the phylogenetic tree of life. Therefore, a deeper look into codon bias in human populations could lead to novel discoveries regarding human evolution. This data is available on the CUBAP Web Portal, allowing users to identify population-specific codon usage biasess in genes and observe their differences across 26 subpopulations.
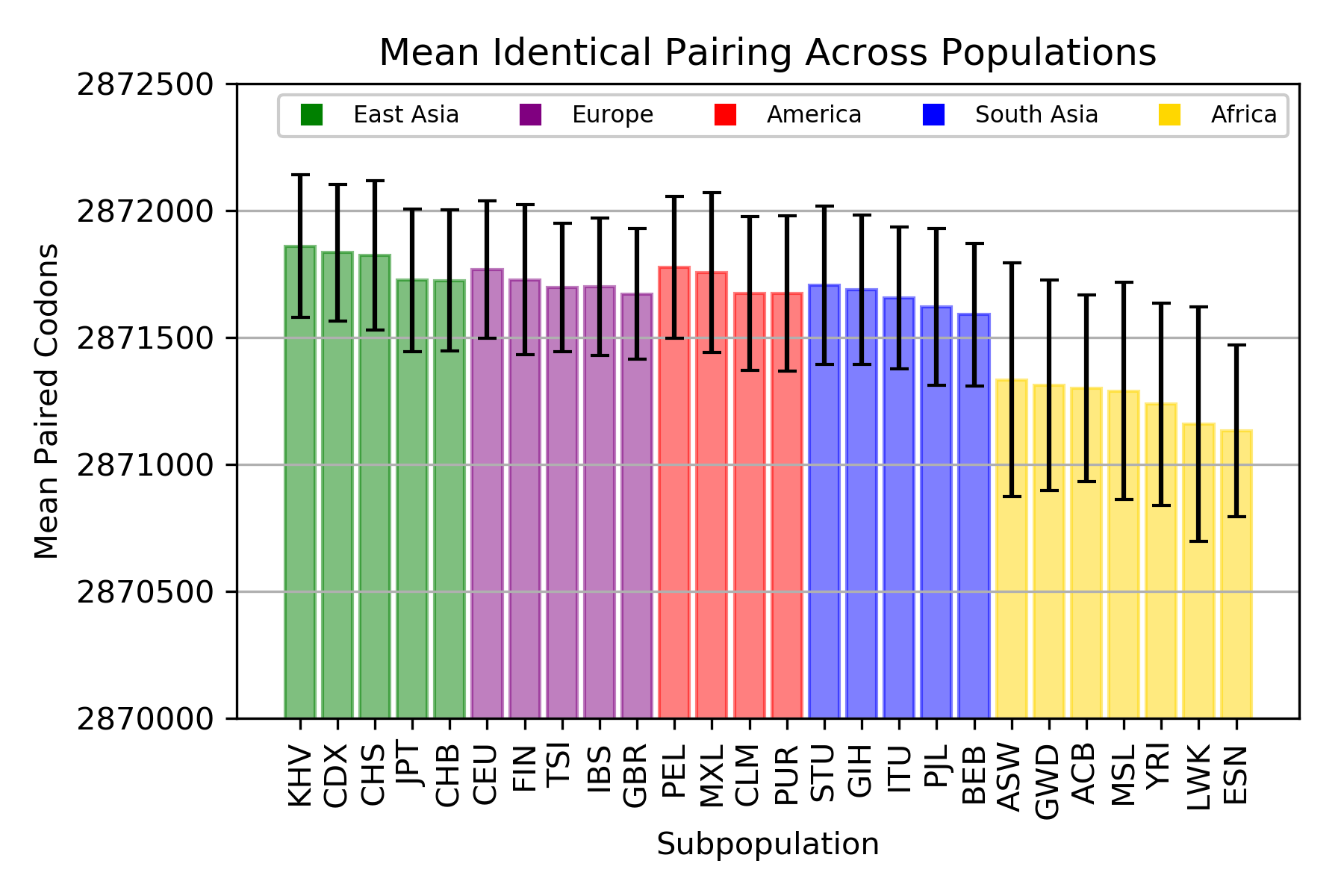
Identical Codon Pairing
ANOVA: F = 45.76828, p = 2.73x10-183
The mean total number of identical codon pairs per individual in each population.
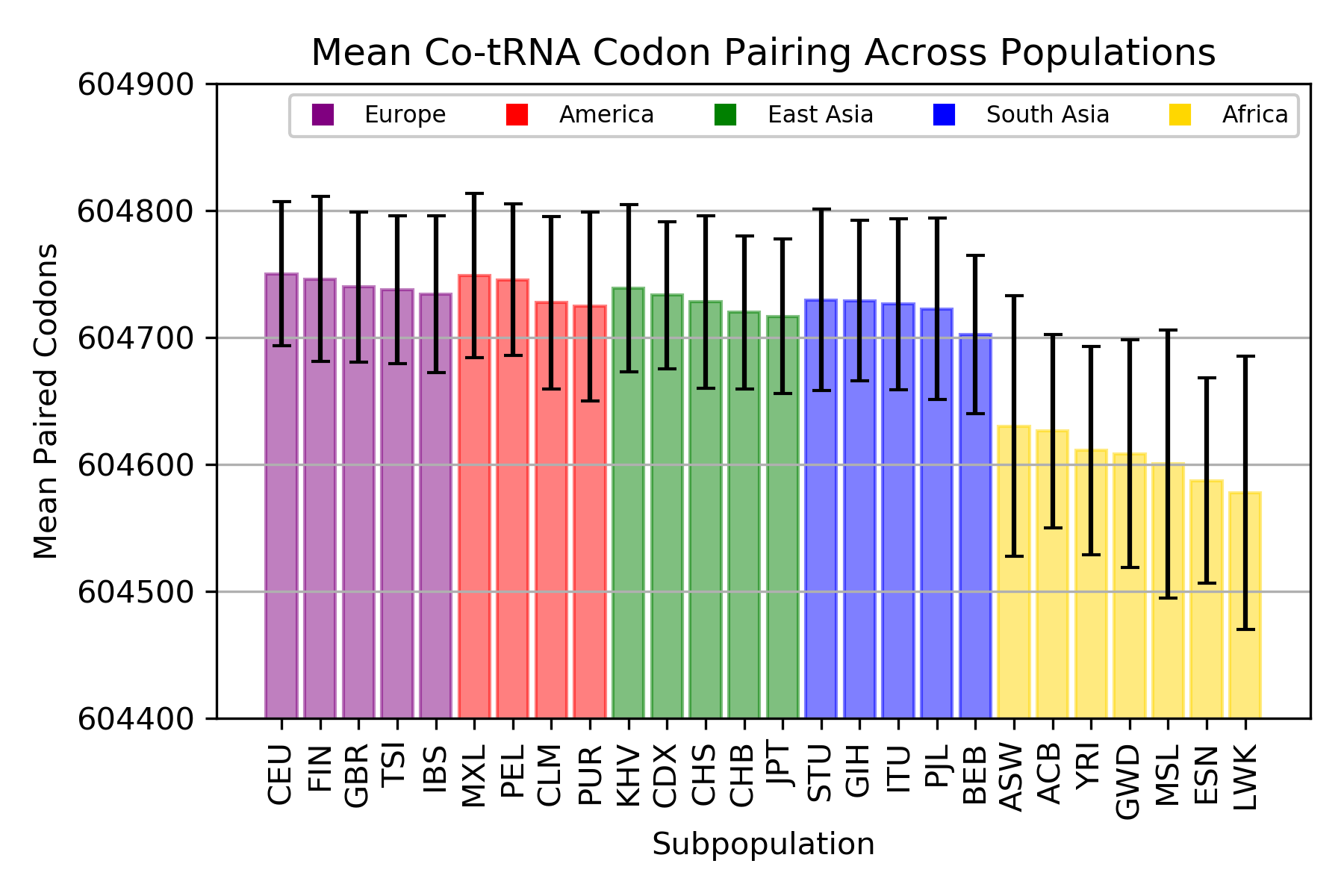
Co-tRNA Codon Pairing
ANOVA: F = 61.88092647, p = 4.7x10-239
The mean total number of co-tRNA codon pairs per individual in each population.
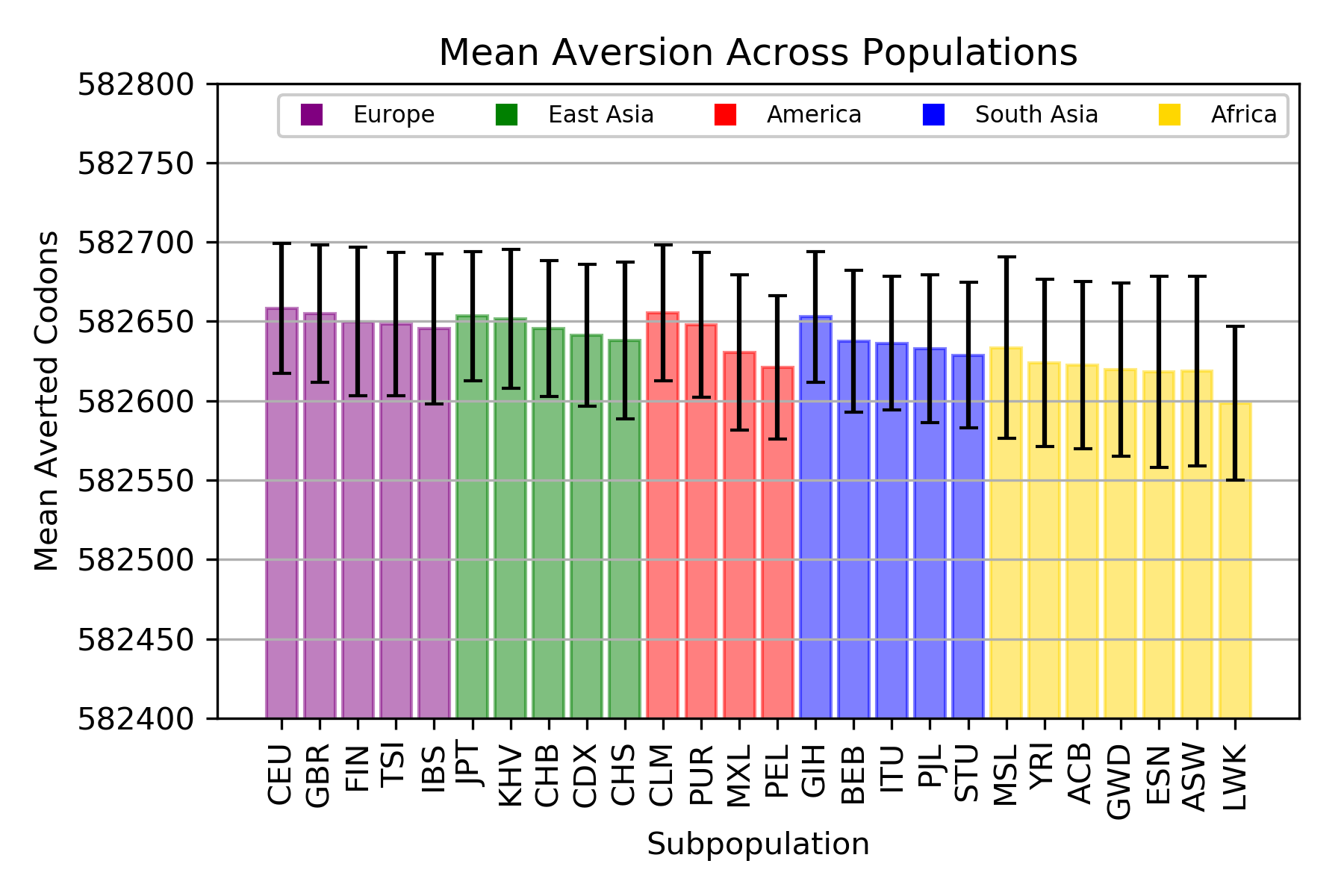
Codon Aversion
ANOVA: F = 9.599947, p = 4.34x10-35
The mean total number of codons averted for individuals in each population.
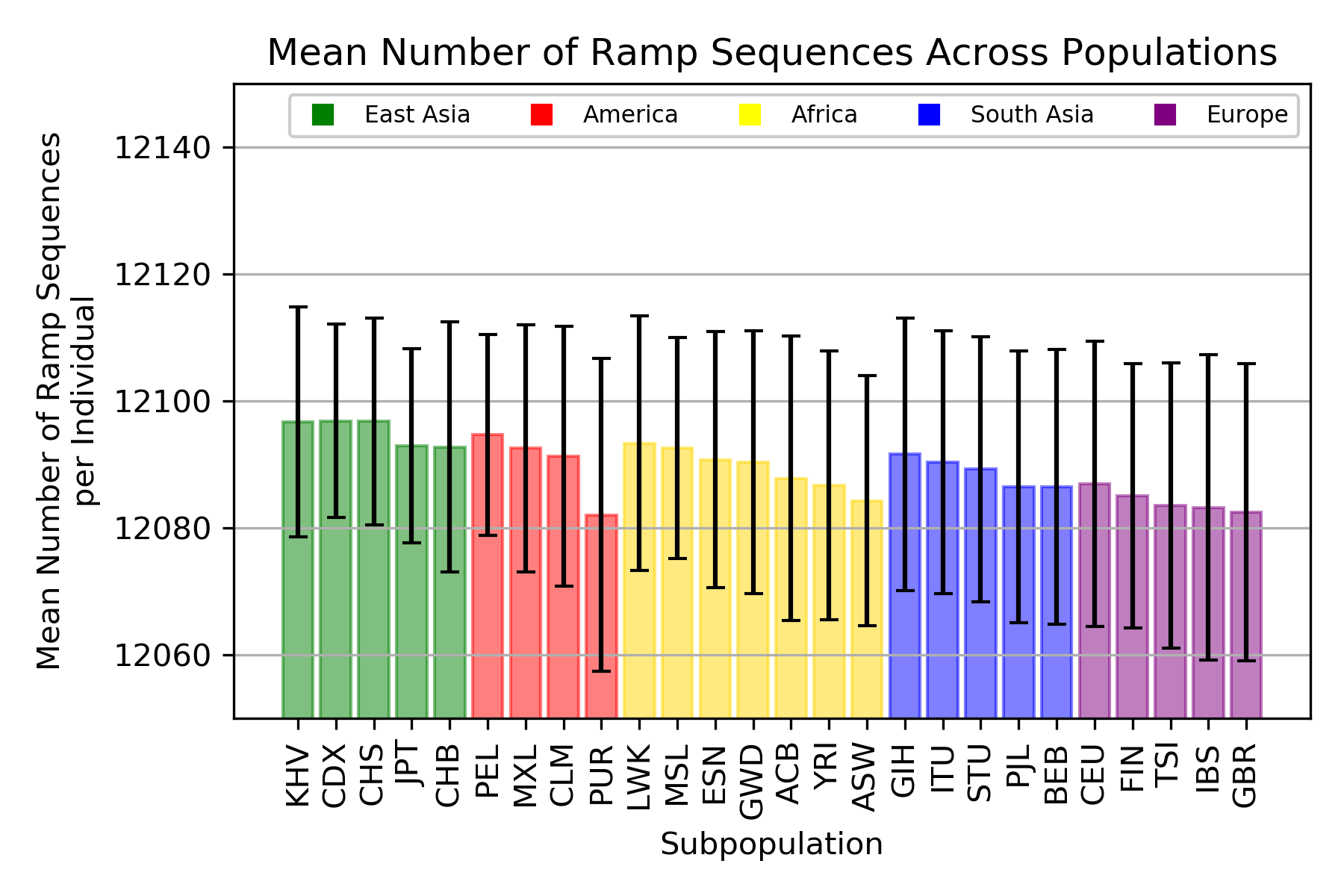
Ramp Sequences
ANOVA: F = 4.693489, p = 1.56x10-13
A ramp sequence is a region of rare codons at the beginning of a gene. These rare codons are translated slowly which allows for proper initation of translation and further accuracy during elongation.
Data
Download the original data files we generated from the 1000 Genomes Project
Codon Frequency and Codon Aversion Data Files
Identical Codon Pairing Data Files
Co-tRNA Codon Pairing Data Files
Ramp Sequence Data File
Nucleotide Composition Data File
Python Scripts